The final draft of the new third edition of the ISO 14155 is being prepared for circulation. This means that at the present stage, only editorial changes are permitted, and therefore this draft version can be used by sponsors designing clinical investigations of medical devices.
Background
The available version has been around since 2011 and has played a significant role in improving the quality of clinical research with medical devices. The standard is accepted by many regulatory authorities globally. One of the reasons for its widely use is that it clearly are describing good practice criteria for clinical use and patient safety. The latest revision of the standard ISO/FDIS 14155:2019 gives even further clarification, consolidation and confidence and contains 10 annexes and three of the annexes are now normative (containing requirements), where we in the previous version had one. The changes implemented are mostly related to the new European Medical Device Regulation (MDR) and the development of more stringent guidelines relevant to the clinical investigations by the European Commission (MEDDEV’s 2.7/1, 2.7/2 and 2.7/3). Additionally, there was a need to further describe study design and align to the guidance of risk management, as the last applies to the design and conduct of the clinical trial. Finally, the GCP standard had also been revised by both the FDA and ICH and even though the GCP cannot directly be transferred to medical device clinical trials the alignment between the GCP and ISO 14155 is desirable to avoid unnecessary confusion.
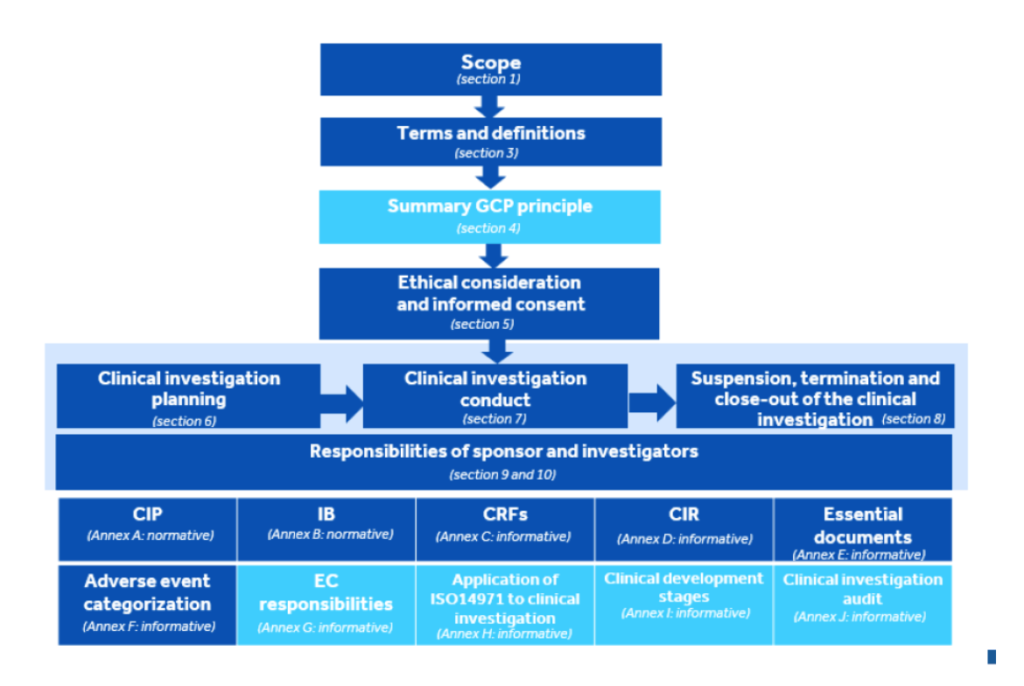
Diagram kindly borrowed form Klaas van’t Klooster, Johnson&Johnson Medical
So, what are the new topics in ISOFDIS 14155:2019?
There are in the Scope of the document a couple of changes to consider beside the listed changes below:
- There is an emphasis on planning evident in a number of key regulatory documents, such as the need for Clinical Evaluation Plan, Risk Management Plan and Biological Evaluation Plan.
- Sponsor should have access to medical expertise relevant to the clinical investigation.
- Recognized that for Software as a Medical Device, not all requirements in the ISO standard might be applicable.
- For IVDs normally the ISO 14155 is not applicable but where a medical device and an IVD are used in an integrated system both the aspects of ISO 14155 and ISO 20916:2019, In vitro diagnostic medical devices – Clinical performance studies using specimens from human subjects – Good study practice, can be applicable to the clinical assessment of the therapy.
Additionally, the following key changes are very nicely described in the foreword of the new standard.
- Inclusion of summary section of GCP principles (Clause 4)
Clause 4 provides specific requirements for risk assessment and study conduct that are indispensable to the fundamental statement at clause 4.c that the right, safety and well-being of human subjects are the most important considerations and prevail over the interest of science and society.
- References to registration of the clinical investigation in a publicly accessible database (Clause 5.4)
It is a requirement that a description of the clinical investigation is entered into a publicly accessible database before the first subject is enrolled and updated throughout the study, and that the results of the study are also disclosed. This is done through clinicaltrials.gov which is US based and for European clinical investigations with the MDR in the European Commission’s medical device database, EUDAMED.
- Inclusion of guidance with regards to clinical quality management (Clause 9.1)
ISO 13485 is not a normative requirement of ISO 14155 and theoretically manufacturers are free to manage product quality any way they want. Therefore, does ISO 14155 contain basic quality assurance requirements, such as for the implementation of written procedures, quality records and auditing. Following the ISO 14155 ensures that the clinical investigation meet recognised quality management criteria so that the results can be used with confidence to conform that the performance and safety requirements specified by the manufacture are fulfilled and that the device meets user needs.
- Inclusion of risk-based monitoring (Clause 6.7)
The description of the content of a monitoring plan has been greatly extended and include a list of 10 pieces of information that must be included in a monitoring plan. It additionally includes a requirement that the monitoring plan must be based, in addition, on the degree of the deviation from the normal clinical practice and the risks assessment (i.e. risk based monitoring).
- Inclusion of guidance on study design and statistical considerations (Annex A)
The access sets out many specific requirements relating to the design of the clinical investigation. Included in this is the point that the study design must always be justified based on a clinical evaluation. Specific requirements for the justification of the statistical design and analysis are also included including recommendations on significance level, sample size justification and statistical methodology.
- Inclusion of guidance for Ethics Committees (Annex G)
An international standard cannot proscribe requirements for public bodies. This standard describes the expected responsibility of the Ethical Committees avoiding inconsistencies and inadequacies in the way ethical aspects of the clinical trials are handled, in particular in relation to informed consent, patient information and vulnerable populations.
- Reinforcement of risk management throughout the process of a clinical investigation (planning to consideration of results,) including Annex H
Annex H contains a flowchart that outlines the process to be followed after identification of any event that suggests that the device or procedure could represent a safety concern. Already in the 2011 version of the ISO 14155 it is required, in order to justify a clinical investigation, residual risks, which are routinely identified as part of a risks analysis conducted according to ISO 14971, as well as risks to the subjects associated with the intended clinical procedure, must be balanced against the anticipated benefits to the subject. ISO 14971 is the only normative reference in ISO 14155, meaning that the requirements of the risk management standard must be applied to all clinical investigations. The risk management principles established by the ISO 14971 must be applied in order to justify the conduct of the clinical investigation and ensure safety of the clinical subjects. Risks arising from the device and the clinical procedure must be evaluated before and throughout the clinical investigation. These same principles must also be applied to the investigation of any serious adverse event, device deficiency or other occurrence that could have an impact on the safety of subjects.
- Clarification of applicability of the requirements of the document to different clinical development stages (Annex I)
Annex I contain a section on applicability of the standards principles, which bring further clarification and provides examples of justifiable exemptions. An important distinction has been introduced between interventional and non-interventional studies, which reflects significant differences in the way post-market Clinical Follow-up studies are regulated. It does although not change the fact that, most part of the standard will apply to post-market studies, event if they are non-interventional.
- Inclusion of guidance on clinical investigation audits (Annex J)
Annex J provides general guidance on the areas that should be examined during the conduct of audits to determine compliance either with the standard or applicable regulations.
In conclusion…
Coupled with the introduction of the MDR this new edition of the soon to come ISO 14155 will give all key stakeholders a more thorough and consistent adoption of the GCP principled within device clinical trials. There are no major surprises or changes required and it is expected that the new version will provide significant improvement and gradually increase patient safety during its implementation.

