FDA’s Compliance Program Manual provides Inspectors with instructions for evaluating industry compliance with regulations and Good Clinical Practice (GCP). Among other activities, the FDA Bioresearch Monitoring (BIMO) Program involves site visits to clinical investigators, sponsors, monitors, contract research organizations, Institutional Review Boards/Ethic Committee (IRB/EC), nonclinical (animal) laboratories, and bioequivalence analytical laboratories. FDA conducts clinical investigator inspections to determine if the clinical investigators are conducting clinical studies in compliance with applicable statutory and regulatory requirements.
In 2001 FDA began the BIMO/OSB (Office of Surveillance and Biometrics) program, which includes the:
- Center for Device & Radiological Health (CDRH)
- Center for Biologics Evaluation & Research (CBER)
Ongoing Investigational New Drug Application(IND)/ or Investigational Device Exemption (IDE) are frequently targeted by FDA, and the selection process is done randomly. Important parameters of studies chosen are time lapse since initiation of the study and the nature of the device; medical product or medical product with a new intended use.
The following topics are the focus of the inspector:
- Organization and Personnel
- Registration of Studies on www.clinicaltrials.gov
- Selection and Monitoring of Clinical Investigators
- Selection of Monitors
- Monitoring Procedures and Activities
- Quality Assurance
- Safety / Adverse Event Reporting
- Data Collection and Handling
- Record Retention
- Financial Disclosure
- Electronic Records and Electronic Signatures
- Test Article
- Emergency Research
- International Data – Human Drugs and Biologics
As of December 20th 2016, the Office of Scientific Investigation (OSI) did 1122 inspections, which is 55 audits more than in 2015. Of those, 639 inspections took place in the US and 373 inspections took place outside the US. The number of inspections performed outside the US has increased. In 2012, only 251 inspections took place outside the US as compared to 373 inspections in 20161; sixty-seven percent of these were done in Europe. Source: 1. https://www.fda.gov/downloads/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDER/UCM438250.pdf
CDHR did altogether 156 (23%) of all inspections, CBER 93 (14%) and Center for Drug Evaluation and Research (CDRH) were accountable for 415 (63%) inspections.
Common European deficiencies are similar to the US inspectional findings and include the following:
- Inadequate monitoring
- Failure to bring investigators into compliance
- Protocol deviations
- Records
- Inadequate investigational product accountability
- Inadequate (S)AE reporting
- Protection of subjects
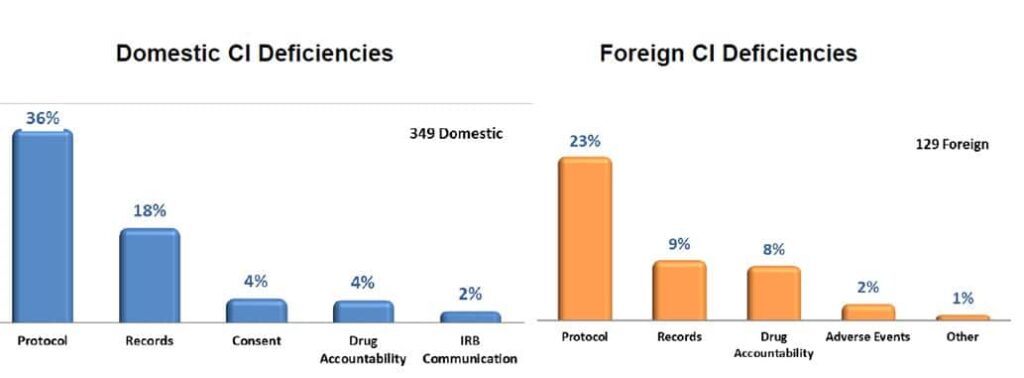
Most Common Clinical Investigation (CI) Deficiencies found during the inspections in both Europe and the US.
- Failure to follow the investigational plan and/or regulations
- Protocol deviations
- Inadequate recordkeeping
- Inadequate accountability for the investigational product
- Inadequate communication with the IRB/EC
- Inadequate subject protection
- Failure to report Adverse Events (AEs)
- Informed consent issues
FDA’s inspection of clinical investigators is not limited to the United States (U.S.). International inspections are generally conducted when the studies are part of a marketing application submitted to FDA and provide data critical to decision-making on product approval. In addition, following final classification of the inspection by the FDA Center, the inspection results are shared with the FDA division assigned to review the marketing submission. The assigned review division considers the results of the inspection during its review of the sponsor’s marketing submission. If the inspection reveals serious violations of FDA’s clinical investigation regulations (e.g., failure to follow the investigational plan), the assigned review division may request additional analyses of the study data or may reject the affected data in certain situations where FDA considers the data unreliable.
How can Qmed help?
Clinical Investigations outside the US are in focus of the FDA, especially if the collected data are used in an IND/IDE application. It is crusial that the investigator and site are ready for an FDA inspection in those cases. At Qmed Consulting we can help you, with our team of trained global proffesionels, to get your investigator and site ready for an FDA inspection. We have the capability to identify any issues that need to be solved prior to an FDA inspection.

